
Tuesday, July 26, 2016 From rOpenSci (https://ropensci.org/blog/2016/07/26/rotl-paper-published/). Except where otherwise noted, content on this site is licensed under the CC-BY license.
We are excited to announce a paper describing rotl, our package for the
Open Tree of Life data, has been
published. The full
citation is:
Michonneau, F., Brown, J. W. and Winter, D. J. (2016), rotl: an R package to interact with the Open Tree of Life data. Methods Ecol Evol. doi: https://doi.org/10.1111/2041-210X.12593
The paper, which is freely available, describes the package and the data it wraps in detail. Rather than rehash the information here, we will use this post to briefly introduce the goals of the package and thank some of the people that helped it come to be.
🔗
What data does Open Tree have and how can rotl help you get it?
The Open Tree of Life combines
knowledge from thousands of scientific studies to produce a single
source of information about the relationships among all species on
earth. In addition to storing the trees and taxonomies that go into this
project, the Open Tree provides a “synthesis tree” that represents this
combined knowledge. The Open Tree data can be accessed via the web page
linked above, and through an API.rotl takes advantage of this API to
give R users the ability to search for phylogenetic information and
import the results into their R sessions. The imported data can then be
used with the growing ecosystem of packages for phylogenetic and
comparative biology in R.
🔗 A small example
Using rotl to get a tree for a set of names is usually a two step
process. You need to start by matching the set of names you have to
records in the Open Tree database, and obtain unique IDs for each one.
The function tnrs_match_names handles this task:
library(rotl)
apes <- c("Pongo", "Pan", "Gorilla", "Hoolock", "Homo")
resolved_names <- tnrs_match_names(apes)
resolved_names
## search_string unique_name approximate_match ott_id is_synonym flags
## 1 pongo Pongo FALSE 417949 FALSE
## 2 pan Pan FALSE 417957 FALSE
## 3 gorilla Gorilla FALSE 417969 FALSE
## 4 hoolock Hoolock FALSE 712902 FALSE
## 5 homo Homo FALSE 770309 FALSE
## number_matches
## 1 1
## 2 1
## 3 1
## 4 1
## 5 1
Once you have a good set of unique IDs, you will want to fetch a tree.
You can use ott_id to extract just the IDs from the table returned by
tnrs_match_names and pass those to tol_induced_subtree to fetch a
tree with theses species in the tips
tr <- tol_induced_subtree(ott_ids=ott_id(resolved_names))
plot(tr)
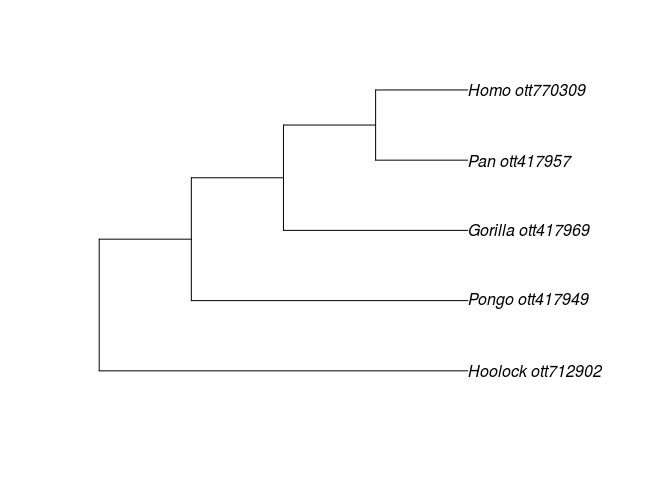
The tree is an object of the ape class phylo, which is used by most
other phylogeny packages in R. There are two vignettes describing how
the fetched trees can be paired with other
data
and used to reproduce a comparative
analysis.
Another
vignette
provides and introduction to the package and includes an FAQ.
🔗 Thanks
We’d like to thank the Open Tree project for providing such an amazing
resource, and for producing a powerful and well-documented API. rotl was
initially developed as part of the Open Tree’s hackathon in Ann Arbor
in
2014.
We’d like to thank the Open Tree project for inviting us to the meeting
and all participants of that meeting for discussions and their feedback
on rotl. The package was reviewed as part of the rOpenSci onboarding
process, and we’d like to thank Scott
Chamberlain for the time and expertise
that went into this review and improved the package.
Filed under
Suggest an edit
Open a pull request
