
Thursday, May 17, 2018 From rOpenSci (https://ropensci.org/blog/2018/05/17/treeio/). Except where otherwise noted, content on this site is licensed under the CC-BY license.
Phylogenetic trees are commonly used to present evolutionary relationships of species. Newick is the de facto format in phylogenetic for representing tree(s). Nexus format incorporates Newick tree text with related information organized into separated units known as blocks. For the R community, we have ape and phylobase packages to import trees from Newick and Nexus formats. However, analysis results (tree + analysis findings) from widely used software packages in this field are not well supported. Some of them are extended from Newick and Nexus (e.g. RevBayes and BEAST outputs), while some of the others are just log files (e.g. r8s and PAML outputs). Parsing these output files is important for interpreting analysis findings.
Information associated with taxon species/strains can be meta-data (e.g. isolation host, time and location, etc.), phenotypic or experimental data. These data as well as analysis finding may be further analyzed in the evolutionary context for downstream integrative and comparative studies. Unfortunately, taxon-associated data are mostly stored in separate files and often in inconsistent formats. To fill this gap, I developed the treeio package for phylogenetic tree data integration.
Currently, treeio is able to read the following file formats: Newick, Nexus, New Hampshire eXtended format (NHX), jplace and Phylip as well as the data outputs from the following analysis programs: BEAST, EPA, HyPhy, MrBayes, PAML, PHYLDOG, pplacer, r8s, RAxML and RevBayes.
🔗 Installation
The treeio package is released within Bioconductor project.
To get the released version from Bioconductor:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("treeio")
Or the development version from GitHub:
## install.packages("devtools")
devtools::install_github("GuangchuangYu/treeio")
🔗 Importing trees with data
🔗 Importing tree data from software outputs
The treeio package provides several parser functions as illustrated in the following table:
| Parser function | Description |
|---|---|
| read.beast | parsing output of BEAST |
| read.codeml | parsing output of CodeML (rst and mlc files) |
| read.codeml_mlc | parsing mlc file (output of CodeML) |
| read.hyphy | parsing output of HYPHY |
| read.jplace | parsing jplace file including output of EPA and pplacer |
| read.mrbayes | parsing output of MrBayes |
| read.newick | parsing newick string, with ability to parse node label as support values |
| read.nhx | parsing NHX file including output of PHYLDOG and RevBayes |
| read.paml_rst | parsing rst file (output of BaseML or CodeML) |
| read.phylip | parsing phylip file (phylip alignment + newick string) |
| read.r8s | parsing output of r8s |
| read.raxml | parsing output of RAxML |
For example, users can parse BEAST output using read.beast function. All the information inferred by by BEAST will be stored in the output object.
file <- system.file("extdata/BEAST", "beast_mcc.tree", package="treeio")
beast <- read.beast(file)
beast
## 'treedata' S4 object that stored information of
## '/Library/R/library/treeio/extdata/BEAST/beast_mcc.tree'.
##
## ...@ phylo:
## Phylogenetic tree with 15 tips and 14 internal nodes.
##
## Tip labels:
## A_1995, B_1996, C_1995, D_1987, E_1996, F_1997, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'height', 'height_0.95_HPD', 'height_median', 'height_range', 'length',
## 'length_0.95_HPD', 'length_median', 'length_range', 'posterior', 'rate',
## 'rate_0.95_HPD', 'rate_median', 'rate_range'.
🔗 Linking external data to phylogeny
In addition to analysis findings that are associated with the tree as we showed above, there is a wide range of heterogeneous data, including phenotypic data, experimental data and clinical data etc., that need to be integrated and linked to phylogeny. For example, in the study of viral evolution, tree nodes may be associated with epidemiological information, such as location, age and subtype. To facilitate data integration, treeio provides full_join method to link external data to phylogeny as demonstrated below:
x <- data_frame(label = as.phylo(beast)$tip.label, trait = rnorm(Ntip(beast)))
full_join(beast, x, by="label")
## 'treedata' S4 object that stored information of
## '/Library/R/library/treeio/extdata/BEAST/beast_mcc.tree'.
##
## ...@ phylo:
## Phylogenetic tree with 15 tips and 14 internal nodes.
##
## Tip labels:
## A_1995, B_1996, C_1995, D_1987, E_1996, F_1997, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'height', 'height_0.95_HPD', 'height_median', 'height_range', 'length',
## 'length_0.95_HPD', 'length_median', 'length_range', 'posterior', 'rate',
## 'rate_0.95_HPD', 'rate_median', 'rate_range', 'trait'.
N <- Nnode2(beast)
y <- data_frame(node = 1:N, fake_trait = rnorm(N), another_trait = runif(N))
full_join(beast, y, by="node")
## 'treedata' S4 object that stored information of
## '/Library/R/library/treeio/extdata/BEAST/beast_mcc.tree'.
##
## ...@ phylo:
## Phylogenetic tree with 15 tips and 14 internal nodes.
##
## Tip labels:
## A_1995, B_1996, C_1995, D_1987, E_1996, F_1997, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'height', 'height_0.95_HPD', 'height_median', 'height_range', 'length',
## 'length_0.95_HPD', 'length_median', 'length_range', 'posterior', 'rate',
## 'rate_0.95_HPD', 'rate_median', 'rate_range', 'fake_trait',
## 'another_trait'.
🔗 Combining tree data
To allow comparative study, treeio supports combining multiple phylogenetic trees into one with their node/branch-specific attribute data.
The following example merges a tree analyzed by BEAST and CODEML.
beast_file <- system.file("examples/MCC_FluA_H3.tree", package="ggtree")
rst_file <- system.file("examples/rst", package="ggtree")
mlc_file <- system.file("examples/mlc", package="ggtree")
beast_tree <- read.beast(beast_file)
codeml_tree <- read.codeml(rst_file, mlc_file)
merged_tree <- merge_tree(beast_tree, codeml_tree)
merged_tree
## 'treedata' S4 object that stored information of
## '/Library/R/library/ggtree/examples/MCC_FluA_H3.tree',
## '/Library/R/library/ggtree/examples/rst',
## '/Library/R/library/ggtree/examples/mlc'.
##
## ...@ phylo:
## Phylogenetic tree with 76 tips and 75 internal nodes.
##
## Tip labels:
## A/Hokkaido/30-1-a/2013, A/New_York/334/2004, A/New_York/463/2005, A/New_York/452/1999, A/New_York/238/2005, A/New_York/523/1998, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'height', 'height_0.95_HPD', 'height_median', 'height_range', 'length',
## 'length_0.95_HPD', 'length_median', 'length_range', 'posterior', 'rate',
## 'rate_0.95_HPD', 'rate_median', 'rate_range', 'subs', 'AA_subs', 't', 'N',
## 'S', 'dN_vs_dS', 'dN', 'dS', 'N_x_dN', 'S_x_dS'.
After merging, the merged_tree object contains the whole set of node/branch attributes from both beast_tree and codeml_tree. The tree object can be converted to tidy data frame using tidytree package and visualized as hexbin scatterplot of dN/dS, dN and dS inferred by CODEML versus rate (substitution rate in unit of substitutions/site/year) inferred by BEAST on the same branches, as an example to demonstrate comparison of node attributes inferred by different software.
library(tidytree)
library(ggplot2)
as_data_frame(merged_tree) %>%
dplyr::select(dN_vs_dS, dN, dS, rate) %>%
subset(dN_vs_dS >=0 & dN_vs_dS <= 1.5) %>%
tidyr::gather(type, value, dN_vs_dS:dS) %>%
ggplot(aes(rate, value)) + geom_hex() +
facet_wrap(~factor(type, levels = c('dN_vs_dS', 'dN', 'dS')),
scale='free_y') +
ylab(NULL)
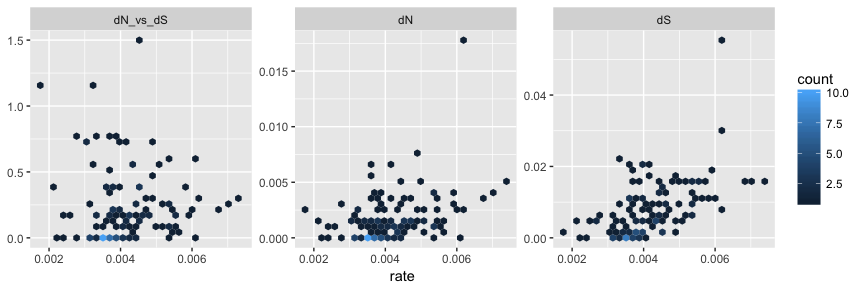
With this feature, users are able to compare/integrate evolutionary inferences obtained from different software packages/models.
🔗 Further readings
For more details of importing trees with data, please refer to the vignette. In addition, treeio also supports exporting tree data to BEAST Nexus format, which allows software output format conversion and also enables combining external data with tree into a single file.
The following screenshot shows an example of exporting CODEML output to BEAST Nexus file. The tree was visualized using FigTree and colored by dN. For more details, please refer to the vignette, exporting trees with data.

All the data parsed/merged by treeio can be converted to tidy data frame using tidytree and can be used to visualize and annotate the tree using ggtree.
For more details, please refer to the online vignettes:
Filed under
Suggest an edit
Open a pull request
