
Tuesday, January 28, 2020 From rOpenSci (https://ropensci.org/blog/2020/01/28/babette/). Except where otherwise noted, content on this site is licensed under the CC-BY license.
babette 1 is a package to work with BEAST2 2, a software platform for Bayesian evolutionary analysis from R.
babette is a spin-off of my own academic research. As a PhD I work on models of diversification: mathematical descriptions of how species form new species. Instead of working on a species' individuals, I work on species as evolutionary lineages. A good way to show the evolutionary relationships between species are phylogenies. For example, this is a phylogeny, with four species, eloquently named ‘1’, ‘2’, ‘3’ and ‘4’:
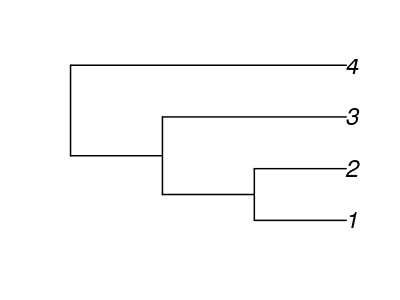
In my research, I simulate thousands of these. I call these phylogenies ’the truth’. Then I wonder, would such a phylogeny actually be true, how well would we be able to infer it? I say ‘infer’, instead of ‘measure’, as we cannot measure phylogenies directly in nature.
In nature, however, we can sequence the DNA of species. From these DNA sequences, we use software to neatly arrange these sequences, creating a DNA alignment. Such DNA alignments are where most researchers start from. Here is such an alignment, of four (eloquently named) species and their aligned DNA sequences, in which a different color represents a different nucleotide (an individual element of a DNA sequence):
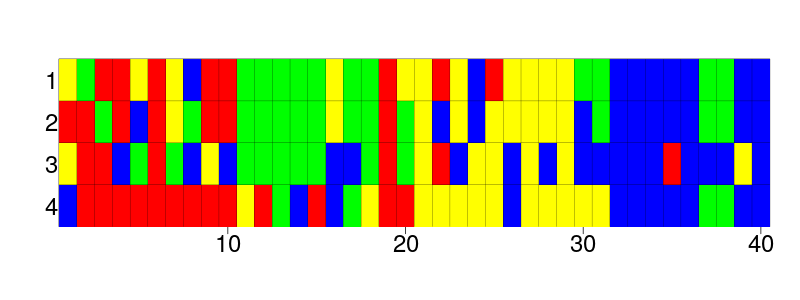
It is hard to only guess by eye which of the four species are most related. Luckily, we have software for such problems, for example, BEAST2. BEAST2 is a Bayesian phylogenetic tool that infers a posterior from a DNA alignment. The posterior contains multiple phylogenies, in which those that are more likely are present more often. Here is what these posterior trees look like when superimposed:

Spoiler: actually, the phylogeny shown was the true phylogeny underlying the alignment! You can see the inference has problems recovering it, as there is no single dominant phylogeny visible, but a mixture of multiple ones instead. Part of my research is to measure exactly the difference between the original phylogeny and those in the posterior. Therefore, I would not only need thousands of phylogenies, but also thousands of posteriors. There was a practical reason to make this go smoothly!
🔗 Before babette
Before babette existed, it would take three steps to plot the phylogenies in a posterior:
| Step | Input | Tool | Output |
|---|---|---|---|
| 1 | alignment | BEAUti | BEAST2 input file |
| 2 | BEAST2 input file | BEAST2 | posterior files |
| 3 | posterior files | DensiTree | plot of the phylogenies |
- Run BEAUti to create a BEAST2 input file
- Run BEAST2 to create a posterior
- Run DensiTree to plot the phylogenies in a posterior
BEAUti (I assume the pun to BEAST is intended) is a beginner-friendly program with a graphical user interface. To analyse an alignment such as above, one needs at least 7 mouse clicks or 3 keyboard commands to create a BEAST2 input file. BEAST2 can be called from the command line to read the input file and create a posterior saved as multiple files. DensiTree is another beginner-friendly program with a graphical user interface to load the phylogenies of a posterior and plot these.
Again, I would need thousands of these posteriors. Doing these steps manually would be a drag and would cost me thousands of mouse clicks. Instead, I created babette to work with BEAST2 from R.
🔗 With babette
We are going to reproduce the workflow described above with babette. First, we’ll load babette into memory:
library(babette)
Again, babette is a package to work with BEAST2 from R. The Bayesian inference of BEAST2 works on an alignment, stored in FASTA format. This is what such a FASTA file looks like:
>1
gcaagagtaacccccgccaggagtaggggcctttttcctt
>2
aacatagcaacccccgccacgtgtgggggtctttttcctt
>3
gaatcactgtcccccttcacgatggtgtgtttttatttgt
>4
taaaaaaaaagactatcgaagggggtgggggtttttcctt
Well spotted, this FASTA file contains the same alignment as the picture above!
Say, we have this alignment stored as a file with the name ‘alignment.fasta’ in our present working directory:
fasta_filename <- "alignment.fasta"
# Check if it really exists
stopifnot(file.exists(fasta_filename))
To obtain a BEAST2 posterior with babette, do:
output <- bbt_run(fasta_filename)
And plotting the posterior trees:
plot_densitree(
output$alignment_trees,
alpha = 0.01,
consensus = as.character(c(1:4)),
cex = 2.0,
scaleX = TRUE,
scale.bar = FALSE
)
Note that for plotting the posterior trees, I’ve added many arguments to make it a beautiful blog post picture :-)
🔗 More about babette
babette actually is a suite of five packages, calling and/or offering an alternative for the BEAST2 tool suite:
| What | BEAST2 suite | babette suite |
|---|---|---|
| Create a BEAST2 input file | BEAUti | beautier |
| Run (only) BEAST2 | beast | beastier |
| Visualize posterior parameter estimates | Tracer | tracerer |
| Visualize posterior phylogenies | DensiTree | babette |
| Install BEAST2 packages | BEAUti | mauricer |
The BEAST2 tool suite has - at the moment - more functionality than babette. The babette GitHub page has a place where feature requests can be and are made. If you want something added and you volunteer to help (that is, answer my naive questions and testing that feature), let me know!
Although babette may not have all BEAST2 functionality, babette already helped me create thousands of posteriors from an R script! Also the analysis done in this blog post, can be found at the babette GitHub repository.
🔗 Why rOpenSci
I enjoy having my academic manuscripts reviewed. We expect the articles we read to be clear and the opinion of our peers help achieve this for our own work. But when I read/run the code in the unreviewed supplementary materials section, there is a loss in clarity. Same goes for R packages written by academics: the article that describes these packages are well-written, but the actual code can be cumbersome to work with. To set a good example, I wanted my code reviewed.
I enjoy having my code reviewed. Even when trying to write exemplary and spotless code, reviewers usually expose the weaker spots in the software. My rOpenSci reviewers did an excellent job at that indeed! Thanks to them, I think babette went from a good package to an excellent one.
🔗 Acknowledgements
Yacine Ben Chehida and Paul van Els supplied the first use cases. Reviews were provided by David Winter and Joëlle Barido-Sottani, with a contribution from Guangchuang Yu. Raphael Scherrer and Jana Riederer helped by submitting bug reports.
🔗 References
Bilderbeek, Richel JC, and Rampal S. Etienne. “babette: BEAUti 2, BEAST 2 and Tracer for R.” Methods in Ecology and Evolution (2018). https://doi.org/10.1111/2041-210X.13032 ↩︎
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C-H., Xie, D., Suchard, MA., Rambaut, A., & Drummond, A. J. (2014). BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Computational Biology, 10(4), e1003537. https://doi.org/10.1371/journal.pcbi.1003537 ↩︎
Filed under
Suggest an edit
Open a pull request
