
Tuesday, March 29, 2022 From rOpenSci (https://ropensci.org/blog/2022/03/29/rsnps-update/). Except where otherwise noted, content on this site is licensed under the CC-BY license.
🔗 TL;DR
rsnps is a package that enables the retrieval of single nucleotide polymorphism (SNP) data from the NCBI’s dbSNP database and openSNP by providing wrappers for the APIs. Single nucleotide polymorphisms represent differences at one specific position in a detected biological sequence compared to the reference.
ncbi_snp_query() now returns all reported variant allele frequencies in dbSNP in column maf_population in form of a tibble. Previously (version <= 0.4.0), it reported only the allele frequency from gnomAD in column maf as a data.frame.
🔗 Changes
The NEWS file will tell you about the changes of rsnps 0.4.0 to 0.5.0, but there is one change that we think will be particularly relevant to users which we will highlight in this blog post.
ncbi_snp_query() is the function that pulls data from NCBI’s dbSNP, a database of single-nucleotide polymorphisms (SNP). This database lets a user query for a SNP of interest and returns a plethora of information, among them genomic position, associated gene, clinical significance, and - relevant for this blogpost - the allele frequency. The allele frequency varies typically (on average) between different populations, sometimes just a little (e.g. rs562556), sometimes a lot (e.g. rs11677783). This is why dbSNP collects allele frequency estimates from different studies and populations.
Until version 0.4.0 ncbi_snp_query() reported the allele frequency estimated from gnomAD. For example, for SNP rs420358 the ncbi_snp_query() output used to look like this:
ncbi_snp_query("rs420358")
#> query chromosome bp class rsid gene
#> 2 rs420358 1 40341239 snv rs420358
#> alleles ancestral_allele variation_allele seqname
#> 2 A,C,G,T A C,G,T NC_000001.11
#> assembly ref_seq minor maf
#> 2 GRCh38.p12 A C 0.8614
We have now changed two things:
ncbi_snp_query()output now includes a newmaf_populationcolumn which contains all reported allele frequencies, not only from one study or population (for backwards compatibility, themafcolumn stays the same).- To do that,
ncbi_snp_query()returns now a tibble (and not a data frame).
🔗 Examples
Let’s have a look at the output of the two SNPs mentioned above. You can see that a) maf_population is a new list-column (<list>), and b) dat is a tibble.
library(rsnps)
(dat <- ncbi_snp_query(c("rs11677783", "rs562556")))
Getting info about the following rsIDs: rs11677783, rs562556
# A tibble: 2 x 16
query chromosome bp class rsid gene alleles ancestral_allele
<chr> <chr> <dbl> <chr> <chr> <chr> <chr> <chr>
1 rs116777… 2 219842264 snv rs116777… "" T,A T
2 rs562556 1 55058564 snv rs562556 "PCSK… G,A G
# … with 8 more variables: variation_allele <chr>, seqname <chr>, hgvs <chr>,
# assembly <chr>, ref_seq <chr>, minor <chr>, maf <dbl>,
# maf_population <list>
🔗 One row per study (“long data”)
Here is a visual to show how much the allele frequencies vary per SNP.
First, we lengthen the data frame, so that each SNP and population/study are on a separate line.
(dat_maf <- dat %>%
select(query, maf_population) %>%
unnest(cols = c(maf_population)))
# A tibble: 43 x 5
query study ref_seq Minor MAF
<chr> <chr> <chr> <chr> <dbl>
1 rs11677783 1000Genomes T A 0.314
2 rs11677783 ALSPAC T A 0.166
3 rs11677783 Estonian T A 0.156
4 rs11677783 GENOME_DK T A 0.1
5 rs11677783 GnomAD T A 0.355
6 rs11677783 GoNL T A 0.164
7 rs11677783 KOREAN T A 0.000342
8 rs11677783 Korea1K T A 0.000546
9 rs11677783 NorthernSweden T A 0.187
10 rs11677783 PRJEB36033 T A 0.3
# … with 33 more rows
Then we display it with the allele frequency on the x-axis and the study along the y-axis.
p1 <- ggplot(data = dat_maf %>% filter(query == "rs11677783") %>% mutate(study = forcats::fct_reorder(study, MAF ))) +
geom_vline(xintercept = c(0, 0.5, 1), linetype = 3, color ="gray") +
geom_point(aes(MAF, study)) +
labs(title = "Allele frequency", subtitle = "rs11677783")
p2 <- ggplot(data = dat_maf %>% filter(query == "rs562556") %>% mutate(study = forcats::fct_reorder(study, MAF ))) +
geom_vline(xintercept = c(0, 0.5, 1), linetype = 3, color ="gray") +
geom_point(aes(MAF, study)) +
labs(title = "Allele frequency", subtitle = "rs562556")
p1 + p2
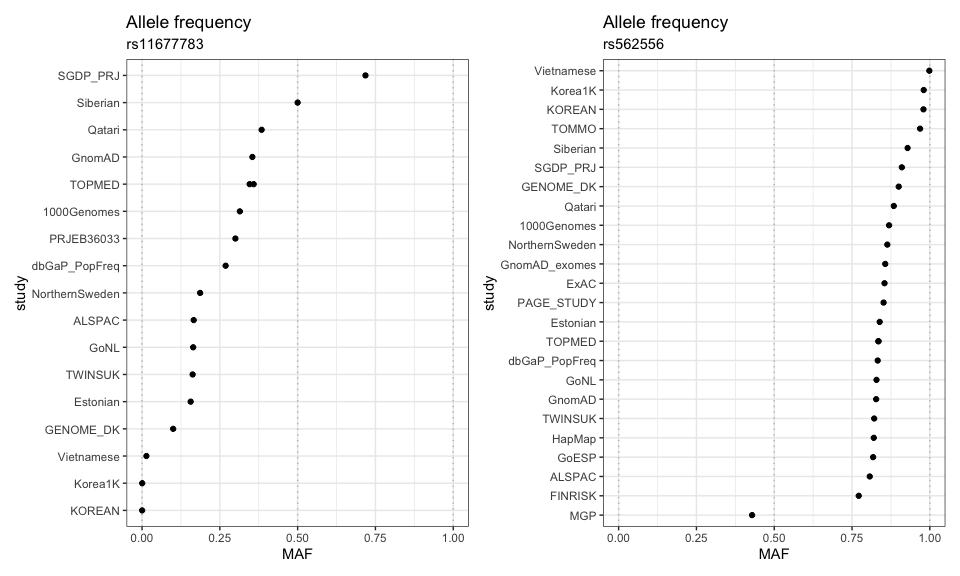
Allele frequencies in dbSNP for rs11677783, rs562556 and various studies/populations.
🔗 One study as column
We can decide to turn the tibble into a data frame again and pick a specific study. For example, here we use the map() function from the purrr package to pull out the study which matches “KOREAN” from the maf_population list. Note that our new column, maf_korean is a dbl, and not a list:
(dat_korean <- dat %>%
mutate(maf_korean = purrr::map(maf_population, ~..1$MAF[..1$study=="KOREAN"])) %>%
unnest(cols = c(maf_korean)))
# A tibble: 2 x 17
query chromosome bp class rsid gene alleles ancestral_allele
<chr> <chr> <dbl> <chr> <chr> <chr> <chr> <chr>
1 rs116777… 2 219842264 snv rs116777… "" T,A T
2 rs562556 1 55058564 snv rs562556 "PCSK… G,A G
# … with 9 more variables: variation_allele <chr>, seqname <chr>, hgvs <chr>,
# assembly <chr>, ref_seq <chr>, minor <chr>, maf <dbl>,
# maf_population <list>, maf_korean <dbl>
🔗 One column per study (“wide data”)
Lastly, we can decide to pivot the study allele frequencies, so that each study has its own column:
dat_maf <- dat %>%
select(query, maf_population) %>%
unnest(cols = c(maf_population)) %>%
select(query, study, MAF) %>%
pivot_wider(values_from = "MAF", names_from = "study", values_fn = min, names_prefix = "maf_") ## if duplicate, picking the minimum
(dat_wide <- left_join(dat, dat_maf, by = "query"))
# A tibble: 2 x 41
query chromosome bp class rsid gene alleles ancestral_allele
<chr> <chr> <dbl> <chr> <chr> <chr> <chr> <chr>
1 rs116777… 2 219842264 snv rs116777… "" T,A T
2 rs562556 1 55058564 snv rs562556 "PCSK… G,A G
# … with 33 more variables: variation_allele <chr>, seqname <chr>, hgvs <chr>,
# assembly <chr>, ref_seq <chr>, minor <chr>, maf <dbl>,
# maf_population <list>, maf_1000Genomes <dbl>, maf_ALSPAC <dbl>,
# maf_Estonian <dbl>, maf_GENOME_DK <dbl>, maf_GnomAD <dbl>, maf_GoNL <dbl>,
# maf_KOREAN <dbl>, maf_Korea1K <dbl>, maf_NorthernSweden <dbl>,
# maf_PRJEB36033 <dbl>, maf_Qatari <dbl>, maf_SGDP_PRJ <dbl>,
# maf_Siberian <dbl>, maf_TOPMED <dbl>, maf_TWINSUK <dbl>,
# maf_Vietnamese <dbl>, maf_dbGaP_PopFreq <dbl>, maf_ExAC <dbl>,
# maf_FINRISK <dbl>, maf_GnomAD_exomes <dbl>, maf_GoESP <dbl>,
# maf_HapMap <dbl>, maf_MGP <dbl>, maf_PAGE_STUDY <dbl>, maf_TOMMO <dbl>
🔗 Conclusion
We have highlighted important changes in ncbi_snp_query() whereby this function now returns a tibble, the column maf_population now returns all allele frequencies available in the NCBI database, and finally maf_population is now a list column instead of a regular column. These improvements which provide additional information and functionality to this package are available in the most recent version of rnsps available on CRAN and on GitHub.
Filed under
Suggest an edit
Open a pull request
