
Monday, December 3, 2018 From rOpenSci (https://ropensci.org/blog/2018/12/03/restez/). Except where otherwise noted, content on this site is licensed under the CC-BY license.
🔗
What is restez?
R packages for interacting with the National Center for Biotechnology Information (NCBI) have, to-date, depended on API query calls via NCBI’s Entrez. For computational analyses that require the automated look-up of reams of biological sequence data, piecemeal querying via bandwith-limited requests is evidently not ideal. These queries are not only slow, but they depend on network connections and the remote server’s consistent behaviour. Additionally, users who make very large requests over extended periods of time run the risk of being blocked.
restez attempts to make large queries to NCBI GenBank more efficient by allowing users to download whole sections of GenBank, create a local database from these downloaded files and then query this mini-GenBank version instead.
This process is far more efficient as the downloaded files are compressed and users can limit the size of the database by only creating it
from sequences of interest (limiting by taxonomic domain and/or sequence size).
restez tries to be user-friendly: a database can be set up in just a few function calls (set path, download and create),
a database can be queried with a consistent set of functions (the gb_*_get() functions),
the number of arguments per function is limited, and the package is designed to integrate with pre-exisiting R packages
that interact with NCBI (rentrez and
phylotaR)
For more a detailed description and for tutorials of the package, please visit the
restez website.
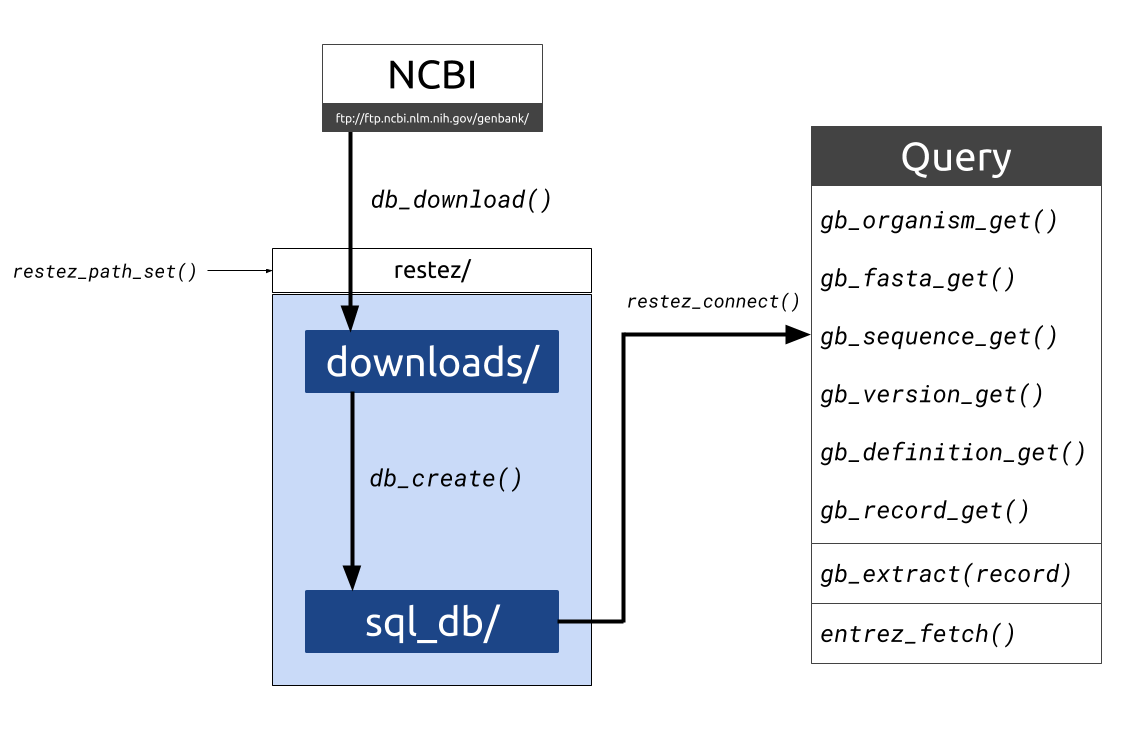 Figure 1. Diagrammatic outline of the
Figure 1. Diagrammatic outline of the restez functions and folder structure. Data is downloaded from NCBI into a file path
set by the user. Raw downloads are stored in “downloads/” the generated database is stored in “sql_db/”. This database can
then be queried with a series of gb_*_get() functions as well as some additional wrappers.
🔗 Installation
restez (v1.0.0) is available from CRAN.
install.packages('restez')
Alternatively, the latest development version can be downloaded from restez’s GitHub page.
devtools::install_github(repo = 'ropensci/restez')
🔗 Usage
To walk us through the basics of restez let’s pretend we’re a microbiologist interested in the sequence diversity among all the bacteriophages that infect Escherichia bacteria. First, we will first need to create our sequence database. Second, we will need to identify the Escherichia phage sequences in this database. And then finally we will need to write out the sequences in a suitable format for a sequence diversity analysis. Thankfully, restez can perform all these steps.
🔗 Set-up
To get started with restez we first have to download and create a database. This set-up consists of three steps:
- set a filepath where we would like to store the database, using
restez_path_set() - download sections of GenBank,
using
db_download() - create the database from the downloaded files,
using
db_create()
Depending on how many GenBank files you select to download, the above process can take up to several hours. In this example, however, we will only download and set up a database for ‘phage’ sequences which should take between 5-10 mintues depending on your machine and internet connection.
🔗 Path
library(restez)
# create a new folder in your working directory to host a database
restez_path <- file.path(getwd(), 'phages')
dir.create(restez_path)
# set the restez path
restez_path_set(restez_path)
The above code will set the restez path. All downloaded files and the created database will be stored in this path.
(Keep a note of the restez path, you will need it later.)
🔗 Download
To download sequences, run the interactive function db_download().
db_download()
This will produce a list of options, like this:
Which sequence file types would you like to download?
Choose from those listed below:
● 1 - 'Bacterial'
542 files and 123 GB
● 2 - 'EST (expressed sequence tag)'
486 files and 243 GB
● 3 - 'Constructed'
372 files and 84.6 GB
● 4 - 'Patent'
340 files and 77.9 GB
● 5 - 'GSS (genome survey sequence)'
308 files and 117 GB
● 6 - 'TSA (transcriptome shotgun assembly)'
234 files and 53.9 GB
● 7 - 'Plant sequence entries (including fungi and algae)'
232 files and 62.8 GB
● 8 - 'HTGS (high throughput genomic sequencing)'
155 files and 36.7 GB
● 9 - 'Invertebrate'
112 files and 24.8 GB
● 10 - 'Environmental sampling'
103 files and 24.1 GB
● 11 - 'Other vertebrate'
94 files and 19.4 GB
● 12 - 'Primate'
59 files and 13.7 GB
● 13 - 'Viral'
58 files and 13.1 GB
● 14 - 'Other mammalian'
55 files and 9.44 GB
● 15 - 'Rodent'
31 files and 7.32 GB
● 16 - 'STS (sequence tagged site)'
20 files and 4.45 GB
● 17 - 'HTC (high throughput cDNA sequencing)'
15 files and 3.43 GB
● 18 - 'Synthetic and chimeric'
10 files and 2.42 GB
● 19 - 'Phage'
5 files and 1.12 GB
● 20 - 'Unannotated'
1 files and 0.00111 GB
Provide one or more numbers separated by spaces.
e.g. to download all Mammalian sequences, type: "12 14 15" followed by Enter
Which files would you like to download?
(Press Esc to quit)
We can download all phage sequences by typing 19. After pressing Enter, we will be told of the likely total file size
of the download. If you have enough free space, push any key to continue. This will then initiate a download process
for all phage sequences files on GenBank.
🔗 Create
After the download process has completed, we can create the database with db_create().
db_create()
This will add all of the downloaded files to the database. It will take a while to complete. When it finishes, we can then identify our Escherichia phage sequences!
🔗 Querying
🔗 Status
After we have built the database, we can query it! For every new R session, we will always need to point restez to the
database using restez_path_set() and then connect to the database with restez_connect(). To get started, let’s see
the database status, is it ready for querying?
library(restez)
restez_path <- file.path(getwd(), 'phages')
restez_path_set(restez_path)
restez_connect()
restez_status()
Checking setup status at ...
────────────────────────────────────────────────────────────────────────────────
Restez path ...
... Path '/phages/restez'
... Does path exist? 'Yes'
────────────────────────────────────────────────────────────────────────────────
Download ...
... Path '/phages/restez/downloads'
... Does path exist? 'Yes'
... N. files 6
... N. GBs 0.34
... GenBank division selections 'Phage'
... GenBank Release 228
... Last updated '2018-11-16 14:46:56'
────────────────────────────────────────────────────────────────────────────────
Database ...
... Path '/phages/restez/sql_db'
... Does path exist? 'Yes'
... N. GBs 1.12
... Is database connected? 'Yes'
... Does the database have data? 'Yes'
... Number of sequences 14911
... Min. sequence length 0
... Max. sequence length Inf
... Last_updated '2018-11-16 14:54:32'
The above status report tells us the database, exists, has data and is connected – which means it’s ready for queries.
(To get a simple TRUE or FALSE for whether the database is ready, use
restez_ready().)
🔗 Get-tools
restez comes with a series of gb_*_get() functions for parsing the GenBank records to pull out specific elements (sequences, definition lines, whole records). We can find records in the database using Accession IDs. To list all Accession IDs in a database, we can use list_db_ids().
# get a random accession ID from the database
id <- sample(list_db_ids(), 1)
# definitions
def <- gb_definition_get(id)[[1]]
print(def)
#> [1] "Unidentified clone B15 DNA sequence from ocean beach sand"
🔗 Escherichia phage sequences
In our scenario, we’re interested in finding and writing out all the Escherichia phage sequences. We can do this by looking up the organism names of the sequence sources of all the sequences in the database. We can then parse these names for "escherichia" and write out the resulting list of sequences.
# get list of ALL IDs in database
ids <- list_db_ids(n = NULL)
# get all organisms
organisms <- gb_organism_get(ids)
# parse for Escherichia
is_escherichia <- grepl('escherichia', organisms, ignore.case = TRUE)
# fetch fasta formatted sequences
fastas <- gb_fasta_get(ids[is_escherichia])
# check ...
cat(fastas[1])
#> >AB000833.1 Bacteriophage Mu DNA for ORF1, sheath protein gpL, ORF2, ORF3, complete cds
#> ACGGTCAGACGTTTGGCCCGACCACCGGGATGAGGCTGACGCAGGTCAGAAATCTTTGTGACGACAACCG
#> TATCAATGCCGGTGTGGTGCTTTACGGCGTTCTGTTCAGTGGCACAACCCCGCTGCCGTCCGTAGTGGAC
#> CTGGATTCGCTGGATGATTACGAGCGTCACTGGCAGACCTGGAAATTCCCGGACGAAACCCCGGAATTTG
#> CCGCACATATCAATGTGAATCAGGAAAAGGATCATGATGCTGAAAATTAAACCCGCAGCGGGAAAAGCCA
#> ....
# write out
write(fastas, file = 'phage_seqs.fasta')
In this little example, we could identify our sequences of interest using restez itself. Ordinarily, however, because sequences can only be looked up via Accession IDs, users will probably not use restez for sequence discovery, only retrieval. For a more adaptable example of searching and fetching sequences, see How to search and fetch sequences
🔗 Integrations
To minimise the coding effort on the part of a user, restez has been built to work with R packages that already connect to
NCBI’s Entrez. After setting up a restez database the same functions of these
other packages can be used to query NCBI Entrez. Internally, restez will query its local database and if it cannot find all
of the requested sequences it will pass these arguments on to these other packages.
For example, users can use the entrez_fetch() function of the rentrez package. Running
this function through restez means a user can first check the local database rather than make lots of queries over the internet.
The function arguments are exactly the same.
Additionally, users can set up up a restez database before launching a phylotaR run.
phylotaR searches NCBI for orthologous sequence clusters for a given taxonomic ID. If a restez database is set-up, phylotaR
will first search the local database before downloading via Entrez.
For more information on these integrations see the additional documentation:
rentrez and restez and
phylotaR and restez
🔗 Future
We have many ideas for improving restez and we welcome forks and pull requests! Our current list of ideas for improvement, include:
- Protein database - the current code could be easily duplicated for working with protein databases, not just GenBank.
- Taxonomy - integration of existing taxonomic packages with
restez. - Retmodes -
restezonly supports text-based return modes, it could be expanded to include XML.
Please see the contributing page for more details and any updates.
If you have any ideas of your own for new features than please open a new issue.
🔗 Acknowledgements
Big thanks to Evan Eskew and Naupaka Zimmerman for reviewing the package; and to Carl Boettiger and Noam Ross for useful comments during the review; and, of course, to Scott Chamberlain for editing!
🔗 Useful Links
🔗 Reference
Bennett, D.J., Hettling, H., Silvestro, D., Vos, R. and Antonelli, A. 2018. 2018. restez: Create and Query a Local Copy of GenBank in R. Journal of Open Source Software, 3(31), 1102, https://doi.org/10.21105/joss.01102
Filed under
Suggest an edit
Open a pull request
